Running time and memory usage #6
Comments
|
It's less dependent on the genome size per se than the fragmentation of the genome itself. If your reference genome is very fragmented (N50 < 10kb etc.) the size of the matrix will be enormous no matter the resolution and it will indeed take up a lot of memory and time. You need to preprocess your initial genome somewhat to avoid this - for instance you could run aggressive filtering so that many smaller contigs are eliminated from the scaffolding and don't clog up your RAM.
What size did you use and did you try generating a matrix using restriction fragments instead? |
|
Hello, It is difficult to give you much information on the speed of the program as it can vary depending on the GPU you have. It is however surprising that it crashed on a 400 GB machine, so there must be some trouble somewhere. Could you please indicate the stats of your input assembly and the exact commands you used to run hicstuff and instagraal? |
|
Thank you for the information, @baudrly and @nadegeguiglielmoni . After comparing with the human sample in your case, I do not think my input assembly is too fragmented with N50=1.05Mb, #contigs=8148. The Hi-C data sets I used for testing had 2*111M reads (file size: 4.5G and 5.1G). Here is the command to generate contact matrix: And for instagraal, all parameters are default. So here is the command I used in the testing program: |
|
It is indeed weird that it crashed on a 400G machine. -What kind of crash was it? (Segfault, MemoryError in python) Do you have a stack trace by any chance? -At what stage did it crash? (While generating the hdf5 files, computing the initial model, in the middle of scaffolding...) -In the meantime, could you try to run the program with a lower resolution (advised for larger genomes) and possibly more filtering, and also with debbuging enabled? Something like:
|
|
Thanks @baudrly ! To be more clear, the contact matrix generated for my 'real sample' is much larger than the 'small sample' because of more replicates of Hi-C reads. Like I mentioned at the beginning,
The 'real sample' crashed after 5 hours with 'out of memory' on the server (400Gig Ram, 6Gig memory on the GPU). The 'small sample' went well, but I terminated it a few days ago because of the endless running...
Sorry, I could not remember or find logs for the 'real sample' because it crashed over one month ago, after which I turned to 'small sample' testing. But what I am sure about is that the program did not manage to build pyramid.
I will check it out and give you feedback. And what impact would it have if I decrease |
Did you concatenate the
To be clear, you don't have to wait for the program to run all the cycles you specified as input, it's only an upper limit. The program creates a new genome after each cycle, so you can check it out and stop if it's satisfactory. Depending on whether you ran the version of the program with the movie enabled, you could have a clearer look into how well the scaffolding did.
Past a certain point adding more cycles only has marginal benefit. In most cases, the scaffolding happens actually pretty quickly, in the first few cycles, as in the example below (each of the twenty frames represents one cycle):
so it's unlikely you'd lose out on much by stopping the program after a while, especially if you can polish the scaffolds afterwards. Also, unless you are really distrustful of the initial contigs' quality, binning more aggressively (level > 5) could speed up the scaffolding by several orders of magnitude at little extra cost, especially given your N50. |
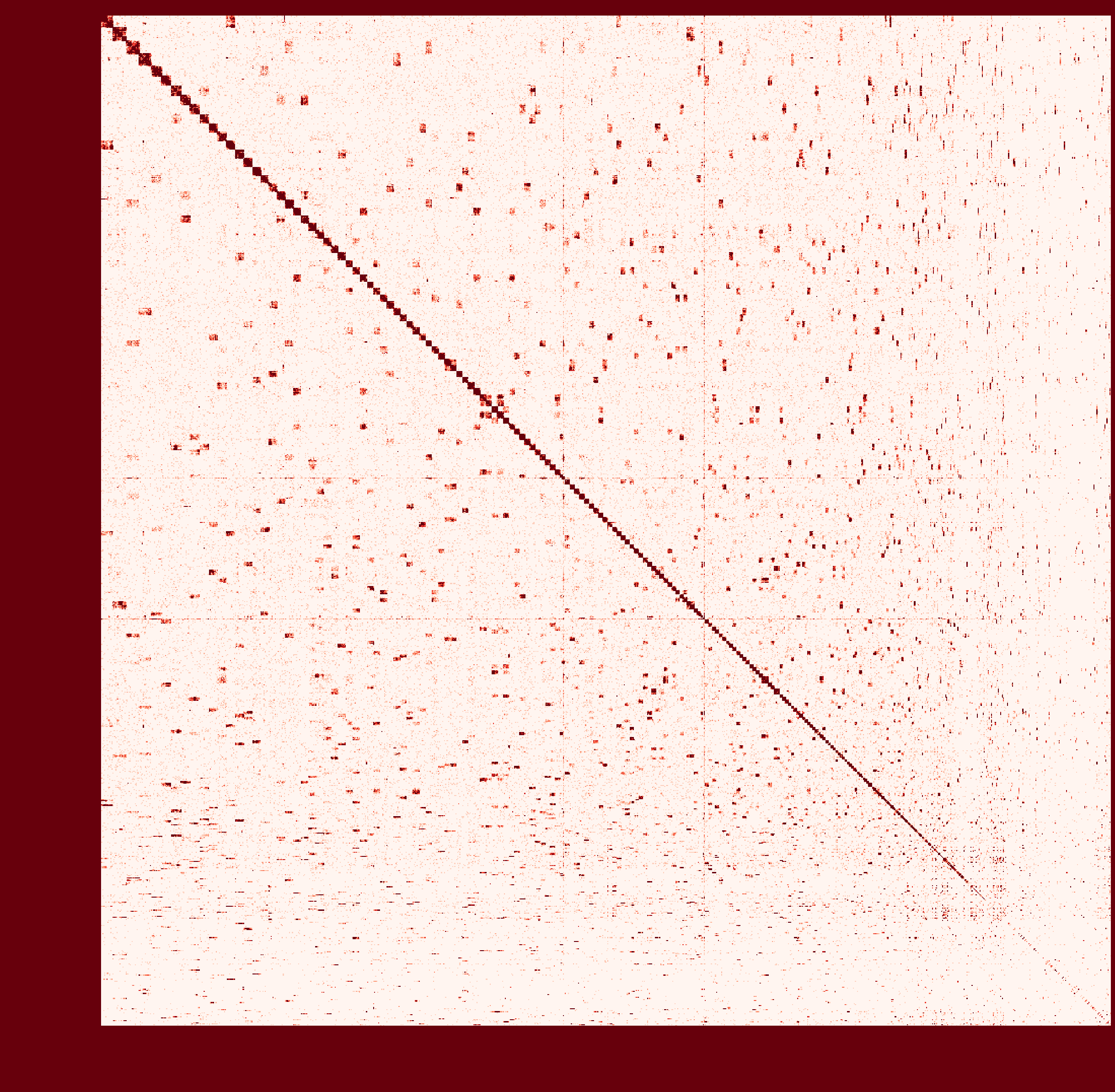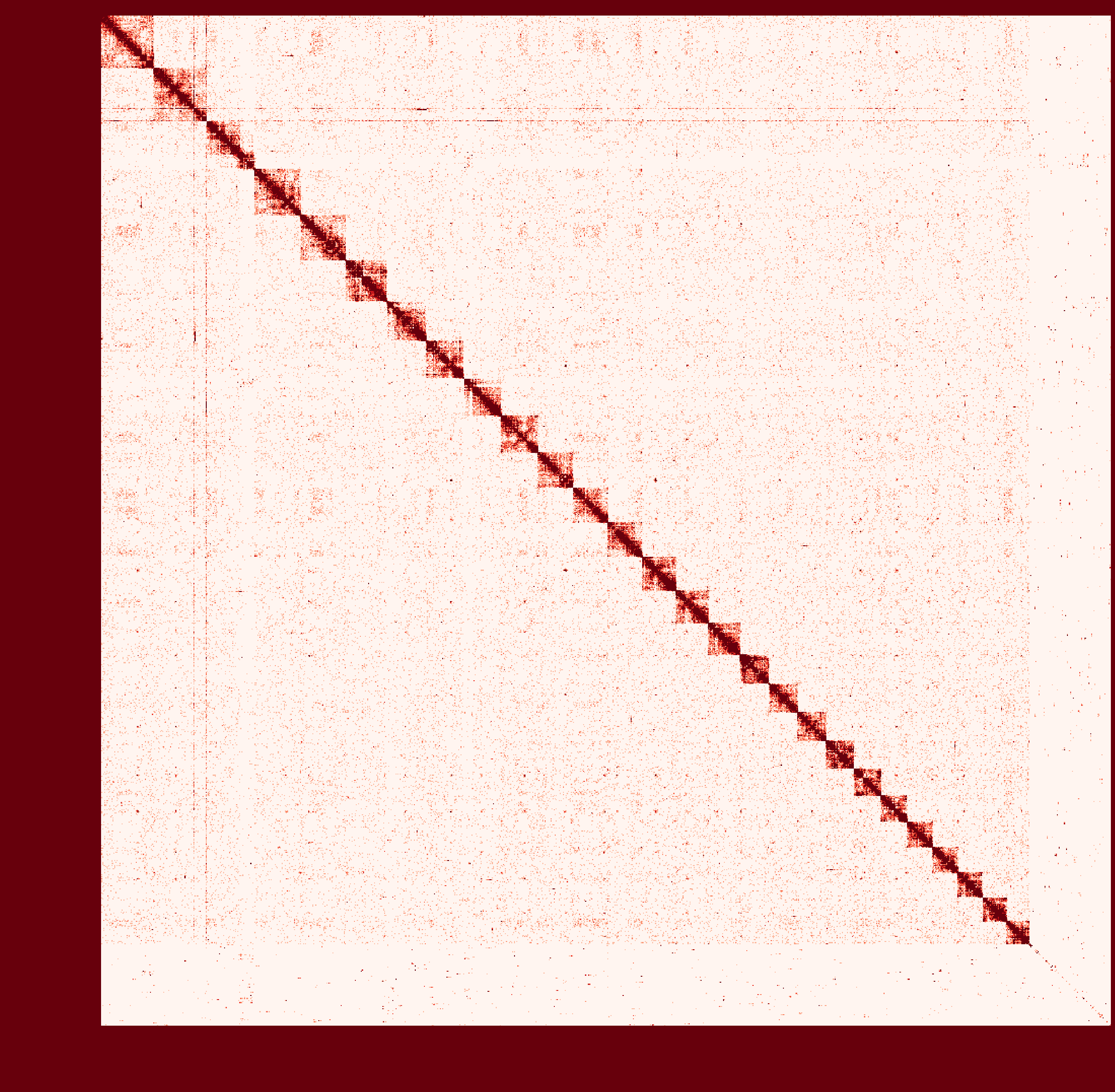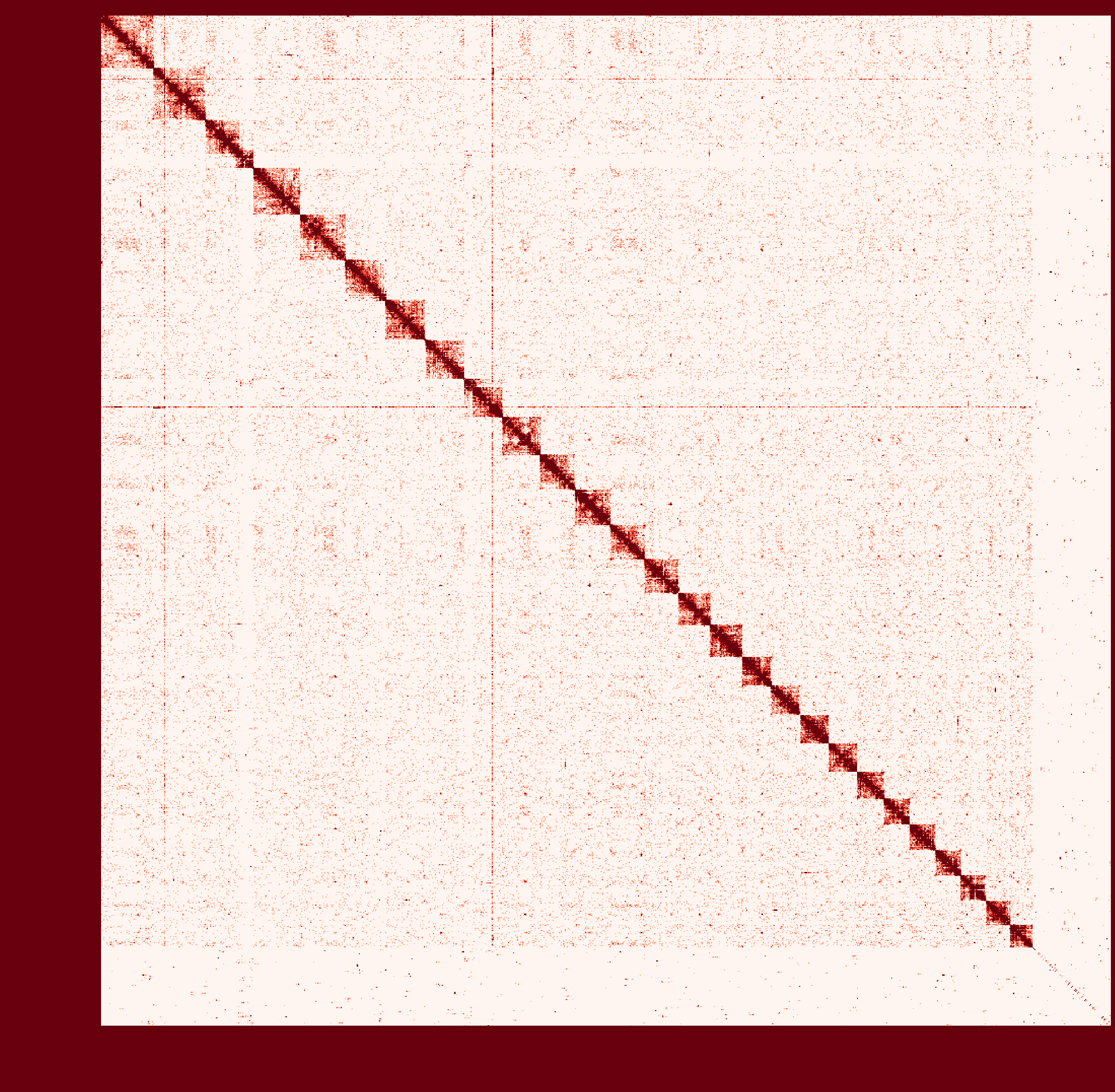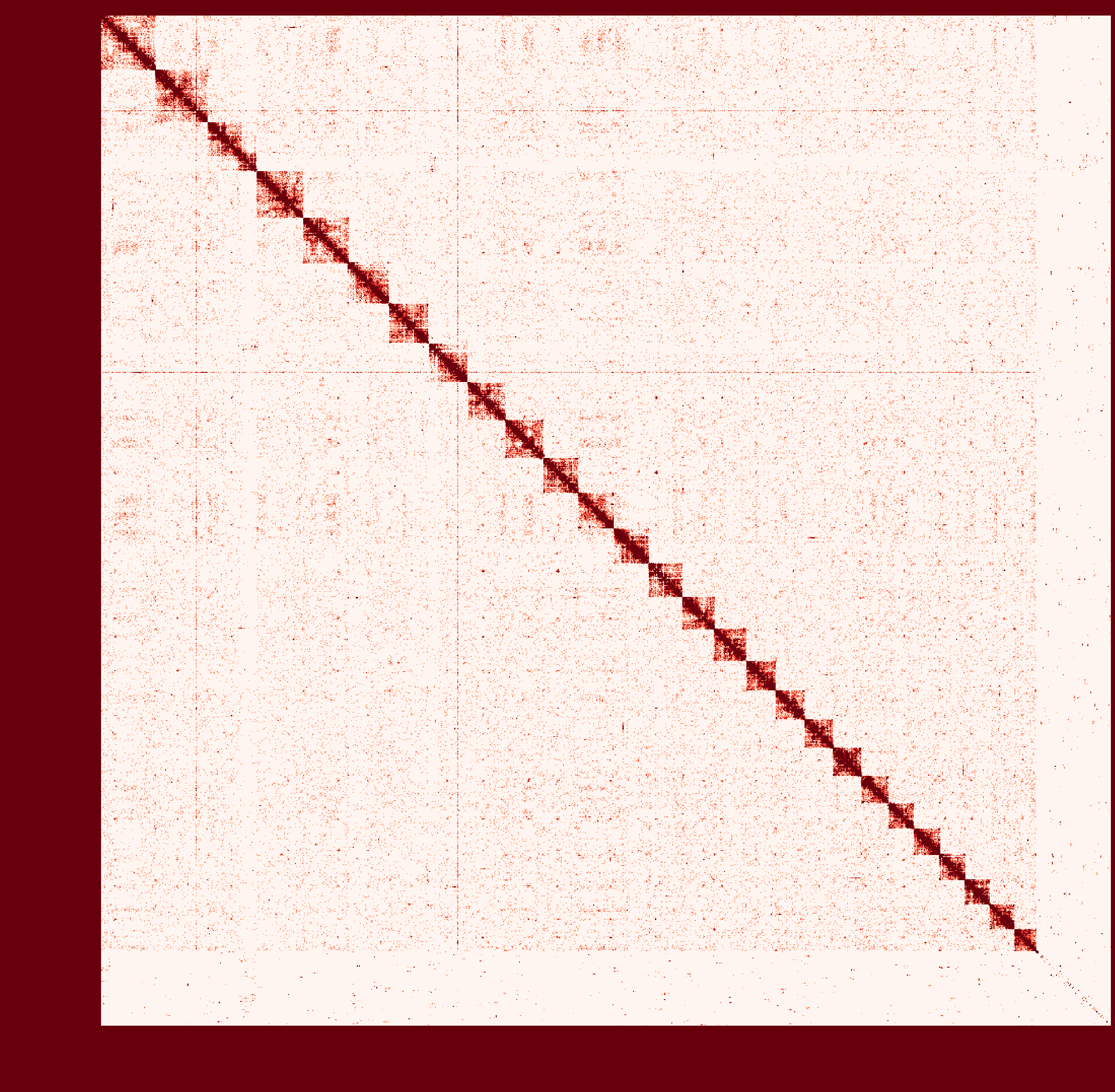
|
A 5.3 GB matrix seems normal, the matrix we had for human with 442 millions was also a little more than 5 GB. But it should not crash a 400 GB machine (in our case, the machine was "only" 128 GB and it did not go over 30 GB to my knowledge). I would suggest that you run again the latest version of hicstuff with all your Hi-C reads, and try instaGRAAL then. |
|
Thank you both for prompt replies!
Sorry, not try it yet. But I will update after running with tuned parameters.
I am curious how you achieved the visualization. Should I run HiC-box to have a look at the dynamics of scaffolding results, or there are parameters to enable? FYI, I use
I used latest version |
If you're talking about a visualization like this: If you're talking about the twenty-frame animation above, I used the
I recommend using a newer version (currently 0.1.6 on PyPI but your best bet is to simply install the very latest from github, possibly this one if you don't want to rely on OpenGL). |
|
To improve speed, you should go for the version without OpenGL because the display slows down the process. |
|
Hello there, Do you have any clues about how to solve this problem? |
|
Hello, I am sorry for not answering earlier. We are investigating this issue. in the meantime, could try installing this fork https://github.com/nowanda92/instaGRAAL ? You may have an error that the kernels are missing somewhere, then you should just copy them from the repo directly to where they are missing. |
|
Hello, I have the same issue with |
|
I tried it with the default parameters, and it runs to: Do you know how many cycle it gonna take? And if the job is killed by the system, is the generated genome.fasta good to use? |
|
@anhong11 By default instaGRAAL runs for 100 cycles. If the job is killed before it reaches 100 cycles, you can use the output as long as the likelihood has converged (you can plot list_likelihood.txt to check that). Rather than using genome.fasta directly, you should run the command instagraal-polish using the output info_frags.txt and the initial fasta file to perform automatic curation of the assembly. |
|
Thank you. It looks converged, right? |

At first I ran a real-case sample with genome size ~2.6Gb and then
instagraalcrashed on a 400Gig machine. So I decreased the size of fragments contacts, which was generated fromhicstuff(I used several replicates of Hi-C read files to generated contacts information at first. After the machine crashed, I only used one replicate to generate smaller contact files). But still, the smaller sample needs ~30Gig Memory on the host, and 2Gig on the GPU, with 780Mabs_fragments_contacts_weighted.txt.In addition, the program ran super slowly that had ran over 1 month before killed. Before ends, it had generated new fasta file in cycles, taking approximately a day per cycle. But that's only a test sample, so I terminated it in cycle 28.
Nevertheless, could you provide any information about the computational resource usage of the program. Like what I have demonstrated, the 'small' sample seems to run endless... I doubt the possibility to apply it on my real sample.
The text was updated successfully, but these errors were encountered: