Home
This package provides a function to simulate a bacterial pangenome in accordance to the neutral model of mutation, as well as the infinitely many genes model for accessory gene gain and loss proceses. It first generates a random ultrametric tree with as many tips as user defines. Then a process of gene gain and loss is simulated mimicking the Infinitely Many Genes (IMG) model, and obtaining at the end of this step a panmatrix describing the gene presence/absence frequencies at the final time (sampling time). As many genes as columns in the final panmatrix are sampled from a reference multi-fasta file, and each one becomes the most recent common ancestor of each gene family at final time. These gene families will have the presence/absence frequency described in the panmatrix, and each individual gene will follow an evolutionary (mutation) path from the time of gene birth, to the final time, at a given constant rate of substitution and a transition matrix. More details of the algorithm are described in the documentation.
First load the package to the R session:
# Load package..
library(simurg)
#> Loading required package: apesimurg provides a single function , simpg(), and some methods associated to the output class. To access the documentation, run:
?simpg
# or
help('simpg')This function needs a reference multi-fasta file from where to sample the genes that would be subjected to mutation, and this package provides one for tutorial purposes only since, depending on running parameters, the user would probably need a bigger one. A well suited reference file would be roary's pan_genome_referebce.fa output file. For this tutorial we will use the attached reference file, which needs to be first decompressed:
wd <- tempdir() # Set the directory where to decompress to /tmp/
tgz <- system.file('extdata', 'ref_tutorial.tar.gz', package = 'simurg')
untar(tarfile = tgz, exdir = wd)
ref <- list.files(path = wd, pattern = 'ref_tutorial[.]fasta$', full.names = TRUE)
ref
#> [1] "/var/folders/cq/v60_yqxs0qjg01c2n2f6yc2w0000gn/T//RtmpoAZnGK/ref_tutorial.fasta"Now we are ready to run a simulation:
set.seed(123)
pg <- simpg(ref = ref, # Reference multi-fasta file
norg = 20, # Number of organisms to sample
ne = 1e11, # Number of generations from MRCA
C = 100, # Core genome size
u = 1e-8, # Probability of gene gain, per generation
v = 1e-11, # Probability of gene loss, per generation
mu = 5e-12, # Probability of substitution, per site, per generation
write_by = 'genome', # Write simulated sequences to files by genome rather by gene family
dir_out = paste(wd, 'dir_out', sep = '/'), # Directory where to write sequences
replace = TRUE, # Permit gene sampling with replacement
force = TRUE, # Force re writing of output directory if exists
verbose = TRUE)
#> Discarded 13 sequences due to presence of "n".
#> Simulating coalescent tree, τ
#> Simulated branch lengths:
#> 0.00443924874241264 0.00337198988823166 0.0086866331229199 0.000232186464031705 0.000468424800783396 0.00301429729883533 0.00345304716720107 0.00186239492195921 0.0413066130959045 0.000530062674228592 0.0223295568375723 0.0133392979905493 0.0100362009834498 0.0179579919555778 0.0125522693929573 0.0849786129738105 0.260533923269216 0.159586805445518 0.590934835374355
#> Number of tips: 20
#> Number of nodes: 19
#> Branch lengths:
#> mean: 0.1234803
#> variance: 0.04335833
#> distribution summary:
#> Min. 1st Qu. Median 3rd Qu. Max.
#> 0.0005300627 0.0165559184 0.0305985386 0.1224112950 1.0110555641
#> No root edge.
#> First ten tip labels: genome16
#> genome6
#> genome11
#> genome8
#> genome19
#> genome20
#> genome7
#> genome1
#> genome15
#> genome2
#> No node labels.
#> Simulating gene gain and loss.
#> IMG parameters:
#> # Core genome size, C = 100
#> # Probability of gene gain, υ = 1e-08
#> # Probability of gene loss, ν = 1e-11
#> # Number of generations, Ne = 1e+11
#> Derived from parameters:
#> # ϴ = 2Ne υ = 2000
#> # ρ = 2Ne ν = 2
#> # Theoretical (expected) MRCA size: C + ϴ / ρ = 1100
#> # Simulated (observed) MRCA size: C + Poi(ϴ / ρ) = 1096
#> Simulating point mutations:
#> # Mutation rate, μ = 5e-12
#> Number and codon position of substitutions branch-wise simulated:
#> List of 3969
#> $ gene1 :List of 38
#> ..$ 21-24: int [1:785] 630 226 464 76 51 328 46 155 75 494 ...
#> ..$ 24-28: int [1:80] 248 314 298 72 495 327 377 282 548 370 ...
#> ..$ 28-36: int [1:62] 84 198 444 414 376 195 402 504 40 575 ...
#> .. [list output truncated]
#> $ gene2 :List of 38
#> ..$ 21-24: int [1:450] 194 41 180 147 269 74 124 116 175 84 ...
#> ..$ 24-28: int [1:47] 323 3 165 180 223 53 286 312 245 255 ...
#> ..$ 28-36: int [1:39] 142 167 58 127 118 174 120 153 167 121 ...
#> .. [list output truncated]
#> $ gene3 :List of 38
#> ..$ 21-24: int [1:179] 98 5 2 24 43 89 11 63 35 56 ...
#> ..$ 24-28: int [1:18] 90 1 15 1 100 71 12 5 136 92 ...
#> ..$ 28-36: int [1:15] 16 80 137 22 128 51 98 7 147 71 ...
#> .. [list output truncated]
#> [list output truncated]
#> Writing sequences at /var/folders/cq/v60_yqxs0qjg01c2n2f6yc2w0000gn/T//RtmpoAZnGK/dir_out
#> Preparing output.
#> Finnish!Now lets inspect the output:
pg
#> Object of class "pangenomeSimulation".
#> Number of sampled organisms: 20
#> Effective Population Size: 1e+11
#> IMG parameters:
#> Coregenome size: 100
#> Probability of gene gain, per generation: υ = 1e-08
#> Probability of gene loss, per generation: ν = 1e-11
#> Substitution parameters:
#> Probability of substitution, per generation: μ = 5e-12
#> Fasta reference file at /private/var/folders/cq/v60_yqxs0qjg01c2n2f6yc2w0000gn/T/RtmpoAZnGK/ref_tutorial.fasta
#> Sequences written at /private/var/folders/cq/v60_yqxs0qjg01c2n2f6yc2w0000gn/T/RtmpoAZnGK/dir_out
class(pg)
#> [1] "pangenomeSimulation"
mode(pg)
#> [1] "list"
length(pg)
#> [1] 5
sapply(pg, class)
#> coalescent gene_list panmatrix substitutions distances
#> "phylo" "array" "matrix" "list" "list"As you see, the class pangenomeSimulation consists in a list of 5 elements.
The first one is the simulated ultrametric tree, of class phylo (ape package). You can, for example, plot it:
plot(pg$coalescent)
The second one is a gene list, showing each gene family members.
head(pg$gene_list, n = 3)
#> $gene1
#> [1] "genome1_gene1" "genome2_gene1" "genome3_gene1" "genome4_gene1"
#> [5] "genome5_gene1" "genome6_gene1" "genome7_gene1" "genome8_gene1"
#> [9] "genome9_gene1" "genome10_gene1" "genome11_gene1" "genome12_gene1"
#> [13] "genome13_gene1" "genome14_gene1" "genome15_gene1" "genome16_gene1"
#> [17] "genome17_gene1" "genome18_gene1" "genome19_gene1" "genome20_gene1"
#> attr(,"name")
#> [1] "1273269.3_01505"
#>
#> $gene10
#> [1] "genome1_gene10" "genome2_gene10" "genome3_gene10"
#> [4] "genome4_gene10" "genome5_gene10" "genome6_gene10"
#> [7] "genome7_gene10" "genome8_gene10" "genome9_gene10"
#> [10] "genome10_gene10" "genome11_gene10" "genome12_gene10"
#> [13] "genome13_gene10" "genome14_gene10" "genome15_gene10"
#> [16] "genome16_gene10" "genome17_gene10" "genome18_gene10"
#> [19] "genome19_gene10" "genome20_gene10"
#> attr(,"name")
#> [1] "1265739.5_01604"
#>
#> $gene100
#> [1] "genome1_gene100" "genome2_gene100" "genome3_gene100"
#> [4] "genome4_gene100" "genome5_gene100" "genome6_gene100"
#> [7] "genome7_gene100" "genome8_gene100" "genome9_gene100"
#> [10] "genome10_gene100" "genome11_gene100" "genome12_gene100"
#> [13] "genome13_gene100" "genome14_gene100" "genome15_gene100"
#> [16] "genome16_gene100" "genome17_gene100" "genome18_gene100"
#> [19] "genome19_gene100" "genome20_gene100"
#> attr(,"name")
#> [1] "593452.3_01050"The third one is the panmatrix. The first columns are core genes, accesory genes are next to them.
# First five core genes:
pg$panmatrix[, 1:5]
#> gene1 gene2 gene3 gene4 gene5
#> genome1 1 1 1 1 1
#> genome2 1 1 1 1 1
#> genome3 1 1 1 1 1
#> genome4 1 1 1 1 1
#> genome5 1 1 1 1 1
#> genome6 1 1 1 1 1
#> genome7 1 1 1 1 1
#> genome8 1 1 1 1 1
#> genome9 1 1 1 1 1
#> genome10 1 1 1 1 1
#> genome11 1 1 1 1 1
#> genome12 1 1 1 1 1
#> genome13 1 1 1 1 1
#> genome14 1 1 1 1 1
#> genome15 1 1 1 1 1
#> genome16 1 1 1 1 1
#> genome17 1 1 1 1 1
#> genome18 1 1 1 1 1
#> genome19 1 1 1 1 1
#> genome20 1 1 1 1 1
# Some accesory genes, for this example:
pg$panmatrix[, 100:105]
#> gene100 gene101 gene102 gene103 gene104 gene105
#> genome1 1 0 1 0 1 0
#> genome2 1 0 1 0 0 1
#> genome3 1 1 1 1 0 0
#> genome4 1 0 1 0 0 1
#> genome5 1 0 1 1 0 1
#> genome6 1 0 1 0 1 0
#> genome7 1 0 1 0 1 0
#> genome8 1 0 1 0 1 0
#> genome9 1 1 1 1 0 1
#> genome10 1 1 1 1 0 1
#> genome11 1 0 1 0 1 0
#> genome12 1 1 1 1 1 1
#> genome13 1 0 1 0 0 1
#> genome14 1 0 1 1 0 1
#> genome15 1 0 1 0 0 1
#> genome16 1 0 1 0 1 0
#> genome17 1 1 1 1 0 1
#> genome18 1 0 0 0 0 1
#> genome19 1 0 1 0 1 0
#> genome20 1 0 1 0 1 0Next field shows each substitution (codon position) for each gene, per branch. This list is an anidated list that shows, for each gene, all the substitutions that took place on each branch. The names of the branches indicates the segments formed by 2 subsequet nodes in the tree (pg$coalescent); i.e. "11-14" is the segment formed between nodes 11 and 14.
str(pg$substitutions, list.len = 3)
#> List of 3969
#> $ gene1 :List of 38
#> ..$ 21-24: int [1:785] 630 226 464 76 51 328 46 155 75 494 ...
#> ..$ 24-28: int [1:80] 248 314 298 72 495 327 377 282 548 370 ...
#> ..$ 28-36: int [1:62] 84 198 444 414 376 195 402 504 40 575 ...
#> .. [list output truncated]
#> $ gene2 :List of 38
#> ..$ 21-24: int [1:450] 194 41 180 147 269 74 124 116 175 84 ...
#> ..$ 24-28: int [1:47] 323 3 165 180 223 53 286 312 245 255 ...
#> ..$ 28-36: int [1:39] 142 167 58 127 118 174 120 153 167 121 ...
#> .. [list output truncated]
#> $ gene3 :List of 38
#> ..$ 21-24: int [1:179] 98 5 2 24 43 89 11 63 35 56 ...
#> ..$ 24-28: int [1:18] 90 1 15 1 100 71 12 5 136 92 ...
#> ..$ 28-36: int [1:15] 16 80 137 22 128 51 98 7 147 71 ...
#> .. [list output truncated]
#> [list output truncated]To see the nodes in the plotted coalescent tree:
plot(pg$coalescent)
nodelabels()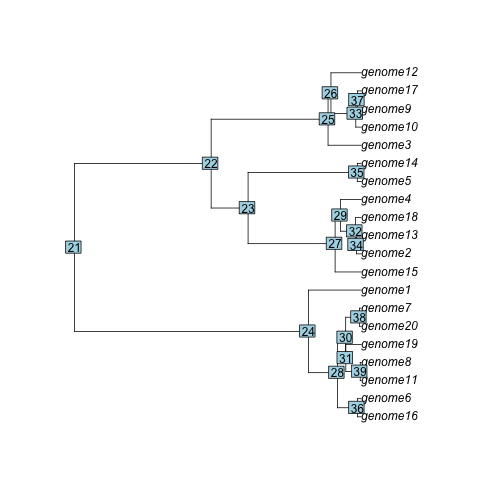
The last field is a list with genetic distances between all pairs of sequences on each gene family.
length(pg$distances)
#> [1] 3969
pg$distances[['gene1']]
#> genome1 genome2 genome3 genome4 genome5
#> genome2 0.491811939
#> genome3 0.465398838 0.322768093
#> genome4 0.497622821 0.060750132 0.324881141
#> genome5 0.484944532 0.267300581 0.311674591 0.276281035
#> genome6 0.148969889 0.500264131 0.464342314 0.503433703 0.487585843
#> genome7 0.152139461 0.502377179 0.467511886 0.506075013 0.489170629
#> genome8 0.144215531 0.498679345 0.462229266 0.502377179 0.484944532
#> genome9 0.471737982 0.342313788 0.091389329 0.343898574 0.320655045
#> genome10 0.470681458 0.340729002 0.090861067 0.343370312 0.319598521
#> genome11 0.142630745 0.499207607 0.461701004 0.502905441 0.484416270
#> genome12 0.469096672 0.332276809 0.087163233 0.331220285 0.314315901
#> genome13 0.491811939 0.017432647 0.326465927 0.069202324 0.269941891
#> genome14 0.483359746 0.267300581 0.313259377 0.275752773 0.014263074
#> genome15 0.495509773 0.086106709 0.320655045 0.089276281 0.270998415
#> genome16 0.146856841 0.498679345 0.465398838 0.500264131 0.486001057
#> genome17 0.470681458 0.337031167 0.087691495 0.338087691 0.315900687
#> genome18 0.494453249 0.020073957 0.325409403 0.064447966 0.274696249
#> genome19 0.144215531 0.494981511 0.460116218 0.499207607 0.484944532
#> genome20 0.150554675 0.501320655 0.468040148 0.505018489 0.488642367
#> genome6 genome7 genome8 genome9 genome10
#> genome2
#> genome3
#> genome4
#> genome5
#> genome6
#> genome7 0.088219757
#> genome8 0.079239303 0.054939250
#> genome9 0.470681458 0.475435816 0.473322768
#> genome10 0.469624934 0.475964078 0.473322768 0.015319599
#> genome11 0.080824089 0.055467512 0.001584786 0.472794506 0.472794506
#> genome12 0.467511886 0.473851030 0.471209720 0.086634971 0.086634971
#> genome13 0.498151083 0.501320655 0.497094559 0.344955098 0.344426836
#> genome14 0.486001057 0.487057581 0.483888008 0.322239831 0.320655045
#> genome15 0.502377179 0.505018489 0.501320655 0.342313788 0.341785526
#> genome16 0.019545695 0.085578447 0.076069731 0.473322768 0.472794506
#> genome17 0.469624934 0.474379292 0.471209720 0.011621764 0.012150026
#> genome18 0.502377179 0.505546751 0.501320655 0.344955098 0.344955098
#> genome19 0.078711041 0.058108822 0.049656630 0.467511886 0.468040148
#> genome20 0.087163233 0.008980454 0.054410988 0.474907554 0.476492340
#> genome11 genome12 genome13 genome14 genome15
#> genome2
#> genome3
#> genome4
#> genome5
#> genome6
#> genome7
#> genome8
#> genome9
#> genome10
#> genome11
#> genome12 0.470681458
#> genome13 0.497622821 0.335446381
#> genome14 0.483359746 0.315900687 0.269413629
#> genome15 0.501848917 0.330692023 0.092974115 0.270998415
#> genome16 0.077654517 0.469096672 0.496566297 0.483888008 0.500264131
#> genome17 0.470681458 0.085050185 0.339672478 0.317485473 0.338615954
#> genome18 0.501848917 0.334918119 0.026941363 0.274696249 0.089276281
#> genome19 0.050184892 0.468568410 0.493924987 0.483359746 0.498151083
#> genome20 0.054939250 0.474379292 0.500264131 0.486529319 0.503961965
#> genome16 genome17 genome18 genome19
#> genome2
#> genome3
#> genome4
#> genome5
#> genome6
#> genome7
#> genome8
#> genome9
#> genome10
#> genome11
#> genome12
#> genome13
#> genome14
#> genome15
#> genome16
#> genome17 0.472266244
#> genome18 0.500264131 0.340200740
#> genome19 0.078182779 0.466455362 0.497622821
#> genome20 0.084521923 0.474907554 0.504490227 0.057052298Finally, sequences are witten at dir_out:
list.files(path = paste(wd, 'dir_out', sep = '/'), full.names = TRUE)
#> [1] "/var/folders/cq/v60_yqxs0qjg01c2n2f6yc2w0000gn/T//RtmpoAZnGK/dir_out/genome1.fasta"
#> [2] "/var/folders/cq/v60_yqxs0qjg01c2n2f6yc2w0000gn/T//RtmpoAZnGK/dir_out/genome10.fasta"
#> [3] "/var/folders/cq/v60_yqxs0qjg01c2n2f6yc2w0000gn/T//RtmpoAZnGK/dir_out/genome11.fasta"
#> [4] "/var/folders/cq/v60_yqxs0qjg01c2n2f6yc2w0000gn/T//RtmpoAZnGK/dir_out/genome12.fasta"
#> [5] "/var/folders/cq/v60_yqxs0qjg01c2n2f6yc2w0000gn/T//RtmpoAZnGK/dir_out/genome13.fasta"
#> [6] "/var/folders/cq/v60_yqxs0qjg01c2n2f6yc2w0000gn/T//RtmpoAZnGK/dir_out/genome14.fasta"
#> [7] "/var/folders/cq/v60_yqxs0qjg01c2n2f6yc2w0000gn/T//RtmpoAZnGK/dir_out/genome15.fasta"
#> [8] "/var/folders/cq/v60_yqxs0qjg01c2n2f6yc2w0000gn/T//RtmpoAZnGK/dir_out/genome16.fasta"
#> [9] "/var/folders/cq/v60_yqxs0qjg01c2n2f6yc2w0000gn/T//RtmpoAZnGK/dir_out/genome17.fasta"
#> [10] "/var/folders/cq/v60_yqxs0qjg01c2n2f6yc2w0000gn/T//RtmpoAZnGK/dir_out/genome18.fasta"
#> [11] "/var/folders/cq/v60_yqxs0qjg01c2n2f6yc2w0000gn/T//RtmpoAZnGK/dir_out/genome19.fasta"
#> [12] "/var/folders/cq/v60_yqxs0qjg01c2n2f6yc2w0000gn/T//RtmpoAZnGK/dir_out/genome2.fasta"
#> [13] "/var/folders/cq/v60_yqxs0qjg01c2n2f6yc2w0000gn/T//RtmpoAZnGK/dir_out/genome20.fasta"
#> [14] "/var/folders/cq/v60_yqxs0qjg01c2n2f6yc2w0000gn/T//RtmpoAZnGK/dir_out/genome3.fasta"
#> [15] "/var/folders/cq/v60_yqxs0qjg01c2n2f6yc2w0000gn/T//RtmpoAZnGK/dir_out/genome4.fasta"
#> [16] "/var/folders/cq/v60_yqxs0qjg01c2n2f6yc2w0000gn/T//RtmpoAZnGK/dir_out/genome5.fasta"
#> [17] "/var/folders/cq/v60_yqxs0qjg01c2n2f6yc2w0000gn/T//RtmpoAZnGK/dir_out/genome6.fasta"
#> [18] "/var/folders/cq/v60_yqxs0qjg01c2n2f6yc2w0000gn/T//RtmpoAZnGK/dir_out/genome7.fasta"
#> [19] "/var/folders/cq/v60_yqxs0qjg01c2n2f6yc2w0000gn/T//RtmpoAZnGK/dir_out/genome8.fasta"
#> [20] "/var/folders/cq/v60_yqxs0qjg01c2n2f6yc2w0000gn/T//RtmpoAZnGK/dir_out/genome9.fasta"The write_by parameter allows to write sequences by gene family rather by genome.
A summary method for class pangenoneSimulation is also provided:
spg <- summary(pg)
#> Summary of object of class pangenomeSimulation:
#>
#> ** Coalescent **
#>
#> Phylogenetic tree: object$coalescent
#>
#> Number of tips: 20
#> Number of nodes: 19
#> Branch lengths:
#> mean: 0.1234803
#> variance: 0.04335833
#> distribution summary:
#> Min. 1st Qu. Median 3rd Qu. Max.
#> 0.0005300627 0.0165559184 0.0305985386 0.1224112950 1.0110555641
#> No root edge.
#> First ten tip labels: genome16
#> genome6
#> genome11
#> genome8
#> genome19
#> genome20
#> genome7
#> genome1
#> genome15
#> genome2
#> No node labels.
#> # Coalescent times [ Expected calculated as 2/(i * (i -1)) ]:
#> 'data.frame': 19 obs. of 2 variables:
#> $ Expected: num 0.00526 0.00585 0.00654 0.00735 0.00833 ...
#> $ Observed: num 0.004439 0.003372 0.008687 0.000232 0.000468 ...
#>
#> ** Infinitely Many Genes Model **
#> Parameters:
#> # Core genome size, C = 100
#> # Probability of gene gain, υ = 1e-08
#> # Probability of gene loss, ν = 1e-11
#> # Number of generations, Ne = 1e+11
#> Derived from parameters:
#> # ϴ = 2Ne υ = 2000
#> # ρ = 2Ne ν = 2
#> # Theoretical MRCA size: C + ϴ / ρ = 1100
#> # Simulated MRCA size: C + Poi(ϴ / ρ) = 1096
#> # Expected pangenome size: E[G] = C + ϴ * sum( 1 / (ρ + 0:19) ) = 5390.717
#> # Observed pangenome size: dim( *$panmatrix )[2] = 3969
#> # Expected average number of genes per genome: E[A] = C + ( ϴ / ρ ) = 1100
#> # Observed number of genes per genome: rowSums( *$panmatrix ) =
#> genome1 genome2 genome3 genome4 genome5 genome6 genome7 genome8
#> 1104 1128 1119 1119 1089 1081 1074 1070
#> genome9 genome10 genome11 genome12 genome13 genome14 genome15 genome16
#> 1117 1114 1067 1097 1120 1083 1115 1080
#> genome17 genome18 genome19 genome20
#> 1118 1109 1053 1072
#> $Gene_family_frequency
#> 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
#> 843 580 171 194 617 157 370 399 52 77 78 152 32 24 31 10 10 28
#> 19 20
#> 23 121
#>
#> ** Evolutionary Genetic Model **
#> Parameters:
#> # Probability of substitution, μ = 5e-12
#> # Number of generations, Ne = 1e+11
#> $Evo_dist
#> 'data.frame': 190 obs. of 4 variables:
#> $ G1 : chr "genome1" "genome1" "genome1" "genome1" ...
#> $ G2 : chr "genome2" "genome3" "genome4" "genome5" ...
#> $ norm_cophenetic: num 1 1 1 1 0.184 ...
#> $ mean_gene_dist : num 0.487 0.481 0.487 0.484 0.156 ...
# Coalescence times (log transform)
brln <- spg$Coal_times
plot(log(brln),
xlab = 'Expected coalescence times (log)',
ylab = 'Simulated coalescence times (log)')
#Expect some dispersion at the begining, but correlation towards the end..
abline(0, 1)
# Gene family frequency
freqs <- spg$Gene_family_frequency
barplot(freqs)
# Evolutionary distance (expected vs observed)
dis <- spg$Evo_dist
head(dis)
#> G1 G2 norm_cophenetic mean_gene_dist
#> 1 genome1 genome2 1.000000 0.4867336
#> 2 genome1 genome3 1.000000 0.4811846
#> 3 genome1 genome4 1.000000 0.4869824
#> 4 genome1 genome5 1.000000 0.4840441
#> 5 genome1 genome6 0.184379 0.1560837
#> 6 genome1 genome7 0.184379 0.1562636
plot(x = dis$norm_cophenetic,
y = dis$mean_gene_dist,
xlim = c(0, 1), xlab = 'Normalized cophenetic distances',
ylim = c(0, 1), ylab = 'Mean genetic distances')
#Expect saturation towards the end..
abline(0, 1)
sessionInfo()
#> R version 3.6.0 (2019-04-26)
#> Platform: x86_64-apple-darwin15.6.0 (64-bit)
#> Running under: macOS Mojave 10.14.6
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/3.6/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/3.6/Resources/lib/libRlapack.dylib
#>
#> locale:
#> [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] simurg_0.1.1 ape_5.3
#>
#> loaded via a namespace (and not attached):
#> [1] igraph_1.2.4.1 Rcpp_1.0.1 knitr_1.23 magrittr_1.5
#> [5] MASS_7.3-51.4 lattice_0.20-38 quadprog_1.5-7 fastmatch_1.1-0
#> [9] stringr_1.4.0 plyr_1.8.4 tools_3.6.0 parallel_3.6.0
#> [13] grid_3.6.0 nlme_3.1-140 xfun_0.8 phangorn_2.5.5
#> [17] seqinr_3.4-5 htmltools_0.3.6 yaml_2.2.0 ade4_1.7-13
#> [21] digest_0.6.19 Matrix_1.2-17 reshape2_1.4.3 evaluate_0.14
#> [25] rmarkdown_1.14 stringi_1.4.3 compiler_3.6.0 pkgconfig_2.0.2